
Dr Vennapusa Sivaranjana Reddy
Associate Professor (Chemistry)
Theoretical and Computational Chemistry
1) ESIPT and White-light emission
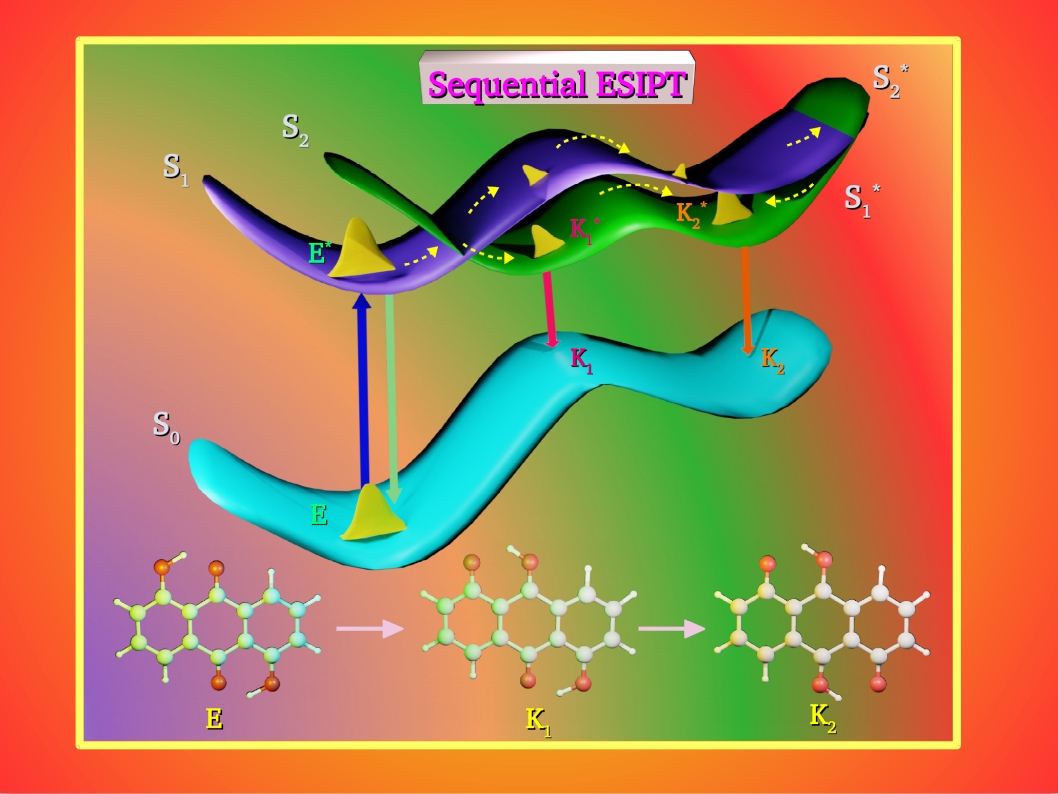
We focus on the study of ultrafast excited state intramolecular proton transfer (ESIPT) in various organic molecules involving 5-membered and 6-membered proton transfer donor-acceptor cycles. Analysis of spectral features associated with ESIPT enol-keto tautomerization is the first step in our study. As the proton transfer (or tautomerization) occurs on the vibrational motion timescales, potential energy surfaces along each of the O-H vibrational modes are explored using computational techniques.Subsequently, quantum molecular simulations on computed potentials provide us the mechanistic pathways and timescales of proton transfer.
2) Ultrafast Intersystem Crossing in Organic Molecules
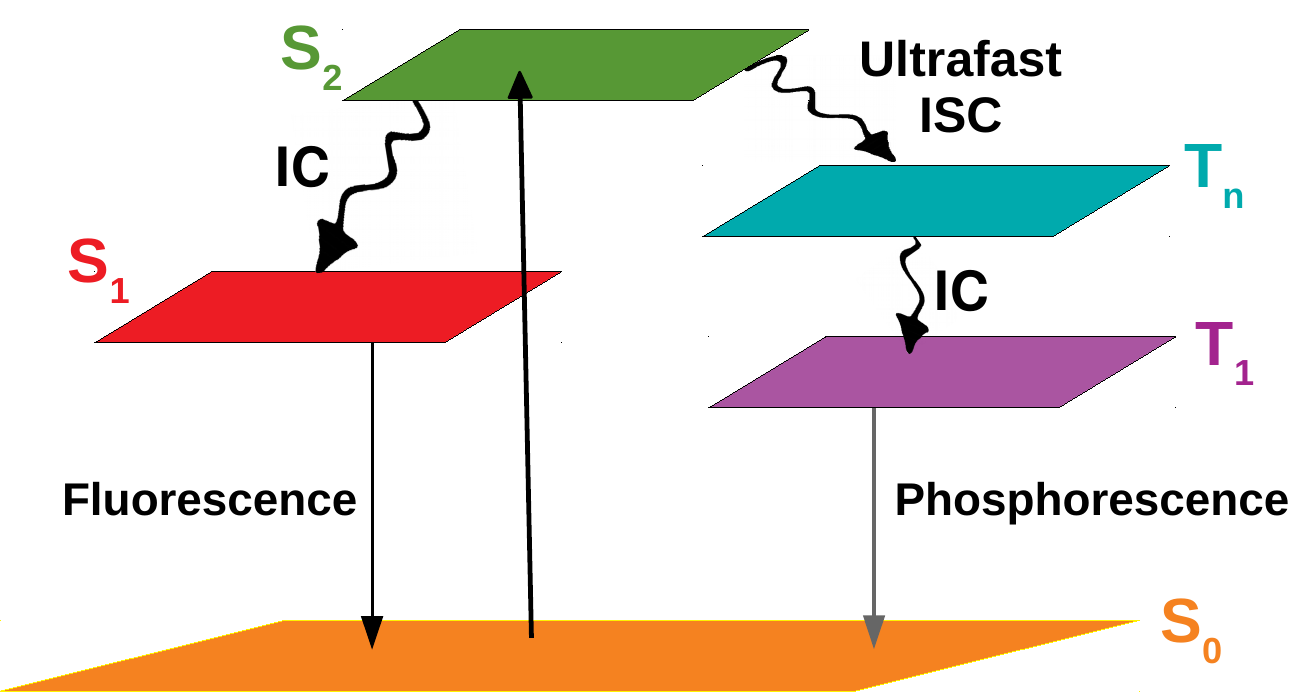
Our interests are to identify the pathways involved in an indirect intersystem crossing in molecular systems. Naphthalene, perylenediimide and pyrene derivatives are the model systems currently under investigation to explore the role of near-degenerate higher singlet-triplet excited states.
3) ESIPT induced triplet formation

Triplet formation pathways in ESIPT tautomers are explored based on the dynamics simulations on PESs generated "on-the-fly". Molecular systems with O-H...S would undergo display ESIPT on a sub-50 fs timescale upon photoexcitation. The proton-transferred tautomer would exhibit mutiple relaxation pathways: (i) nonradiative decay to its ground-state (ii) ultrafast ISC to close-lying higher triplet state of the tautomer form, which ultimately decays into T1 and (iii) reverse ISC. These interesting features of ESIPT are of highly relevant for designing efficient molecules for optoelectronics and triplet sensitization.